
相关热词搜索: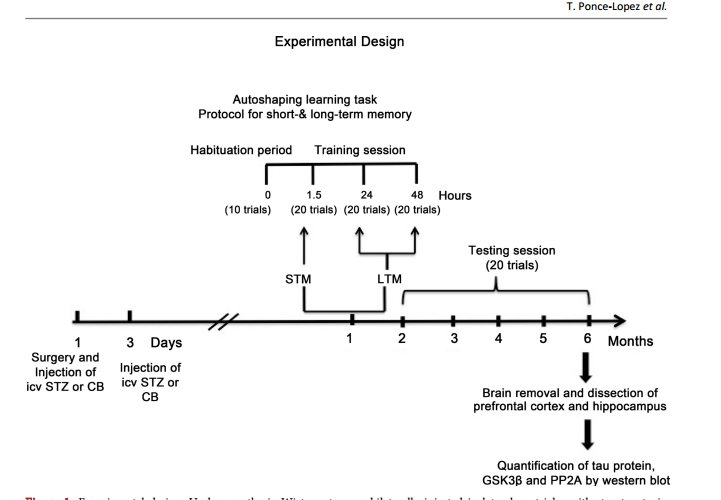 Role of GSK3β and PP2A on Regulation of Tau Phosphorylation in Hippocampus and Memory Impairment in ICV-STZ Animal Model of Alzheimer’s Disease Teresa Ponce-Lopez1,2*, Enrique Hong1, Manuel Abascal-Díaz2, Alfredo Meneses1 1 Deparment of Pharmacobiology, CINVESTAV, México City, México 2 Faculty of Health Sciences, Anahuac University, Huixquilucan State of México, Mexico Abstract Intracerebroventricular administration (ICV) of streptozotocin (STZ) in rats has been associated to desensitization of the insulin receptor (IR) and biochemical changes similar to those occurring in Alzheimer’s disease (AD) or older brains, so it has been proposed as a suitable model for studying some of the pathological features of AD sporadic type (SAD). In this study, we investigated the role of glycogen synthase kinase 3β (GSK3β) and protein phosphatase 2A (PP2A) in the regulation of the phosphorylation of tau (p-tau). Results showed that ICV-STZ treated rats had deficits in short- (1.5-h) and long-term (24- and 48-h) memory after one month of ICV-STZ treatment and six months relative to control rats. The memory deficit was associated to increasing [F(3, 12) = 31.48, p < 0.0001] p-tau in the hippocampus but not in prefrontal cortex (PFC). Likewise, STZ reduced phosphorylation of GSK3β (p-GSK3β) and PP2A in hippocampus and PFC, indicating that GSK3β and PP2A contributed to regulation of p-tau. These data supporting the model with ICV-STZ in rat are adequate to study the progressive memory impairment associated to hyperphosphorylation of tau and the cascade of insulin receptor signaling; confirm that phosphatidyl-inositol-3 kinase-protein kinase B (PI3K-PKB/AktGSK3β) and PP2A are involved in the modulation of proteins responsible for the regulation of neurodegeneration in AD. Keywords Memory Deficit, Tau Hyperphosphorylation, GSK3β, PP2A, Streptozotocin, Hippocampus How to cite this paper: Ponce-Lopez, T., Hong, E., Abascal-Díaz, M. and Meneses, A. (2017) Role of GSK3β and PP2A on Regulation of Tau Phosphorylation in Hippocampus and Memory Impairment in ICVSTZ Animal Model of Alzheimer’s Disease. Advances in Alzheimer’s Disease, 6, 13-31. https://doi.org/10.4236/aad.2017.61002 Received: January 21, 2017 Accepted: March 7, 2017 Published: March 10, 2017 Copyright © 2017 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/ Open Access T. Ponce-Lopez et al. 14 1. Introduction Alzheimer’s disease (AD) is the leading cause of dementia and is characterized by progressive memory loss and a gradual decline in cognitive function, eventually leading to premature death of the inpidual, which occurs typically 3 - 9 years after diagnosis [1]. The neuropathological hallmarks of AD are extracellular senile plaques containing aggregates of the β-amyloid peptide (Aβ), and intraneuronal neurofibrillary tangles (NFTs) [2] [3] [4] that consist mainly of intracellular and abnormally phosphorylated tau protein [3] [5]. These pathologic features are accompanied by decreased synaptic density, which eventually leads to widespread neurodegeneration, loss of synapses and failure of neurotransmitter pathways, particularly those of the basal forebrain cholinergic system, especially in the hippocampus and cortex [3] [6]. Tau phosphorylation (ptau) is regulated by numerous Ser/Thr and phosphatases, including glycogen synthase kinase 3β (GSK3β) and protein phosphatase 2A (PP2A); both are considered to be the major kinase and phosphatase in vivo [7] [8] [9]. Several evidences have been associated between insulin dysfunction and AD [10]-[16]. For instance, hyperphosphorylation of tau protein has been associated with brain insulin deficiency or disorder of insulin signal transduction [17]. A post-mortem study showed that the hippocampal formation in AD cases without diabetes, exhibited markedly reduced receptor/insulin receptor substrate1/ phosphatidyli-inositol-3 kinase (IR-IRS1-PI3K) signaling pathway [18]. The other study showed that peripheral hyperinsulinemia correlated with an abnormal removal of Aβ and an increase in tau hyperphosphorylation, consequently of increased GSK3β activity [19]. In fact, hyperinsulinemia and Type II diabetes mellitus are considered risk factors for SAD [16] [20] [21] [22] [23]. Importantly, diabetes animal models suggest that peripheral insulin signaling dysfunction plays a key role in modulating AD pathology. Indeed, streptozotocin (STZ) administration (200 mg/kg i.p.) produced increase of tau phosphorylation in the brain of non-transgenic mice [7]. Likewise, the ICV administration of low STZ doses (1 - 3 mg/kg) reproduces aspects of SAD abnormalities, including decreased glucose utilization in rat cortical regions and hippocampus [24] [25] [26] [27], cholinergic deficits [25], increase in oxidative stress [28] [29] [30], decrease of IR expression and hyperphosphorylated tau protein in the hippocampus [31], and amyloid formation in leptomeningeal vessels [30]. All these changes are associated to memory impairment, and tau pathology, resulting in central insulin dysfunction [30] [31] [32] [33]. Therefore, herein we are testing that ICV-STZ animal model provides key information about the role of brain insulin disruption in AD pathology, hence, studying the effect at six months following icv STZ administration on memory function, phosphorylated levels of tau and GSK3β, and PP2A levels. 2. Methods 2.1. Animals Male Wistar 4-month-old rats weighing 320 - 340 g were used. The animals were T. Ponce-Lopez et al. 15 placed on an inpidual cage after STZ administration during recovery time after surgery (one week). During the experimental phase they were maintained at 22˚C ± 2˚C and 12 h light/dark cycle, with free access to food and water previous to experimental manipulation. The experimental protocol was revised and approved by the Institutional Review Committee (CICUAL; Project No. 047/02) for the use of animal subjects in compliance with the National Institutes of Health Guide for Care and Use of Laboratory Animals (publication No. 85 - 23, revised 1985). 2.2. Surgical Procedure and Icv STZ Administration Adult male Wistar rats were anesthetized with ketamine (75 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.). The animal body was placed in position in the stereotaxic apparatus and in its head a midline sagittal incision was made in the scalp. Then, holes were drilled in the skull on both sides over the lateral ventricles. The following coordinates were used for ICV injection: 0.8 mm posterior to Bregma, 1.5 mm lateral to sagittal suture, 3.6 mm ventral from the surface of the brain [28]. Coordinates for placement of cannulas were determined by using the atlas ofPaxinos & Watson [34]. STZ was dissolved in citrate buffer (CB; pH 4.5; 28, 32) just prior to injection. The STZ group was injected bilaterally with STZ (3 mg/kg) in two pided doses on Days 1 and 3 asreported previously [28]. The concentration of STZ was adjusted so as to deliver 2 μL/ventricule of the solution as described previously [32]. In control group were given icv injection of the same volume of CB on 1 and 3 day as in STZ injected rats. Post-operatively, as above mentioned rats were maintained in an inpidual cage and received free access to food and water. One week previous to the behavioral task body weights were reduced to 85% by gradually reducing food intake until the last experimental day in order to perform the behavioral task. 2.3. Behavioral Protocol 2.3.1. Autoshaping Learning Task In an autoshaping or sign-tracking setting, a hungry animal is placed in a conditioning chamber to find food pellets (unconditioned stimulus [US]) in the foodmagazine and is then given a Pavlovian sequential pairing (stimulus-stimulus [S-S]) of a lighted key or a retractable-illuminated lever (conditioned stimulus [CS]) and food (US). After a number of such presentations, the animal approaches the CS and presents instrumental responses (conditioned response [CR]), such as peck, nose-poke, and lever-press responses. Then, CR or autoshaped responses result from the S-S association and are sustained by response-stimulus (R-S) association [35]. Importantly, within the continued progress of behavioral memory tasks development, a Pavlovian/Instrumental autoshaping (P/I-A) task combines both Pavlovian and instrumental conditioning; which offers the opportunity to study hippocampus-mediated declarative memory and striatum-mediated R-S “habit formation” [36]. Furthermore, except for magazine training, P/IA is almost completely automatized, considerably reducing human intervention. T. Ponce-Lopez et al. 16 It is sensitive to small increases or decreases in various behavioral parameters (i.e., not measuring the same event twice), including sign tracking (i.e., conditioned behavior directed toward the localized retractable and illuminated lever; CS), and goal tracking (i.e., the place where the US is delivered). The latest is quite important, as it allows the study of bidirectional expression of an enhanced or impaired memory formation. P/I-A clearly separates training for testing sessions, and it has been useful to detect changes in memory formation elicited by drugs or aging [35]. 2.3.2. Food Magazine and Autoshaping Training Following one month of the icv STZ administration, inpidually, each rat was placed in an experimental chamber for a habituation period (≈15 min) with access to 50 food-pellets (45 mg each) previously placed into the food magazine. The criterion was that once the animal ate all 50 food-pellets and presented 150 nose-pokes (as measured by a photocell) into the food-magazine, the autoshaping training program was initiated [35] [37]. The autoshaping program had been reported previously [37] [38], and this consisted of discrete trials. A trial began with the presentation of a retractable and illuminated lever for 8 s (conditioned stimulus; CS) followed by a food-pellet (unconditioned stimulus; US) delivery. There was an inter-trial interval time (ITI) of 60 s. When the animal pressed the CS, it was considered a conditioned response (CR), which shortened the trial, retracted the lever, turned off the light, and a US was delivered. The CR increment or decrement was considered an index for enhancement or impairment in memory consolidation, respectively. There was an autoshaping training session (10 trials) lasting nearly 12 min, and three training/testing (20 trails each) sessions, lasting nearly 24 min. All sessions were conducted over three consecutive days. The autoshaping training session is followed by consecutive training/testing sessions at 1.5 h for short-term memory (STM), and 24 and 48 h for long-term memory (LTM). Subsequently, a 20 minute testing session was conducted each month for 6 consecutive months (Figure 1). 2.4. Measured of Brain Proteins 2.4.1. Tissue Preparation As previously reported [38], the rats treated with CB or STZ were sacrificed by decapitation after the autoshaping testing session at 6 months (Figure 1), and their brains were quickly removed, placed on ice in order to dissect the prefrontal cortex and hippocampus (from CA1 area to dentate gyrus) for each group according to Paxinos and Watson, 2005. Hippocampal and prefrontal cortex were homogenized with lysis buffer containing 150 mM NaCl, 50 mMTris-HCl, 5 mM EDTA and protease inhibitors (PMSF, aprotinin, leupeptin and pepstatin). The lysates were centrifuged at 12,000 g per 20 min at 4˚C. The supernatants were removed and kept in new Eppendorf tubes. Samples were frozen and stored at −70˚C until further analysis. Total protein concentration was determined. The amount of protein was assayed according to the method of [39] (Sigma; Cat. No. B6916). For calibration curve bovine serum albumin (BSA) standard was used (Sigma). T. Ponce-Lopez et al. 17 Figure 1. Experimental design. Under anesthesia Wistar rats were bilaterally injected in lateral ventricles with streptozotocin (STZ) or citrate buffer (CB; vehicle) twice on day 1 and 3, after one month short- (STM) and long-term memory (LTM) was assessed in autoshaping, an associative learning task. Subsequently, testing session was conducted each month for 6 consecutive months. The animals were sacrificed at 6 months, the brains were removed and the hippocampus and prefrontal cortex were dissected in order to measure tau protein, GSK3β and PP2A by western blot. ICV: intracerebroventricular. The concentration was determined by measurement of the absorbance at 595 nm. 2.4.2. Western Blot Equal amounts of total protein (20 μg per sample for enzyme for analyses) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis using 10% polyacrylamide gels and transferred to Polyvinylidenedifluoride (PVDF) membrane [40] [41]. The PVDF membranes were blocked by incubation in 5% non-fat milk added to phosphate buffered saline (PBS-T) containing 137 mM de NaCl, 2.7 mM de KCl, 10 mM de Na2HPO4, 2 mM de KH2PO4, pH 7.4, 0.5% Tween 20, 1 h at 22˚C for p-GSK3β and total GSK3β. Blocked blots were incubated on the next day with primary antibody anti-phospho-GSK3β (Ser9) rabbit (1:1000; Cell Signaling, Inc.), total GSK3β (27 C10) rabbit (1:1000; Cell Signaling, Inc.), Tau (Tau46) Mouse (1:10,000; Cell Signaling, Inc.), phospho-tau (Ser396) (PHF13) Mouse; (1:1000, Cell Signaling, Inc.), PP2A subunit A (6G3) rat (1:1000, Cell Signaling, Inc.) and anti-GAPDH clone 6C5 (1:5000; Milipore, Inc.) overnight at 4˚C. After incubation, the membranes were washed three times T. Ponce-Lopez et al. 18 with PBS-T 1% and incubated for 1 h at room temperature with secondary antibody solution anti-rabbit IgG (1:2000) p-GSK3β, total GSK3β, total tau, phospho-tau and PP2A (1:2000; Milipore, Inc.), and anti-mouse IgG (1:10,000, Milipore, Inc.) for GAPDH. The specificity of the signal was checked on control membranes that were not incubated with primary antibody. After washing three times in PBS, the membranes were immunostained using chemiluminescence western blotting detection reagents (Bio Rad) and exposure to an X-ray film. Relative optical density of bands was analyzed using MCID gel analysis software, Imaging Research Inc. 2.4.3. Statistical Analysis The values of the conditioned responses (CR) were expressed as a percentage of the total trials (10 or 20) per session (mean ± SEM) in the autoshaping test, meaning that, e.g., 2 - 3 CR corresponded to 20% - 30%. The CR was expressed as means (±SEM) and was analyzed by Student’s t test (two groups). The n per group was 6 - 8 animals. The p-tau, total tau, p-GSK3β and total GSK3β values were expressed as means (±SEM) and they were analyzed by one way ANOVA (Three or more groups) followed by Tukey test post-hoc. The PP2A was expressed as means (±SEM) and they were analyzed by Student’s t test (two groups). In all comparisons p < 0.05 was considered as significant. The n per group was 4 - 5 animals. The statistical software used was GraphPad Prism version 6.00 for Macintosh, San Diego California USA. 3. Results 3.1. Short and Long Term Memory Deficit in Icv STZ-Treated Rats The results showed that one month after icv injection of STZ did not change conditioned responses (CR) during phase training. Nevertheless, CR significantly t(18) = 3.128, p = 0.0058 decreased during STM (1.5 h) and t(18) = 3.16, p = 0.0054; t(18) = 4.514, p = 0.0003 LTM (24 & 48 h) relative to vehicle animals (Figure 2(a)). Memory was assessed every month for a period of six months, and the memory deficit was significantly maintained t(14) = 8.804, p < 0.0001; t(14) = 2.98, p = 0.0099; t(14) = 5.688, p < 0.0001; t(10) = 5.179, p = 0.0004; t(10) = 5.440, p = 0.0008; t(12) = 4.371, p = 0.0009, respectively (Figure 2(b)). 3.2. STZ Effect on Tau Phosphorylation (P-Tau) Western blot analysis (Figure 3(a)) demonstrated that p-tau levels were significantly [F(3, 12) = 31.48, p < 0.0001] higher in STZ groups than the control group at 6 months in the hippocampus (Figure 3(b)). There were no changes in total tau levels (Figure 3(b)). The ratio between p-tau/tau was significantly t(4) = 2.456, p = 0.0494 increased (Figure 3(c)) in STZ Western blot analysis (Figure 4(a)) in the PFC p-tau levels significantly [F(3, 12) = 21.01, p < 0.0001] augmented in STZ groups compared with controls at 6 months (Figure 4(b)). However, total tau levels significantly [F(3, 8) =9.154, P < 0.0058] also increased in STZ group compared to control (Figure 4(b)). With T. Ponce-Lopez et al. 19 respect to the ratio between p-tau/tau total was not observed significant differences in control and STZ rats (Figure 4(c)). 3.3. STZ Effect on GSK3β Phosphorylation (p-GSK3β) Western blot analysis (Figure 5(a)) showed the values of p-GSK3β levels significantly [F(3, 12) = 31.81, p < 0.0001] decreased in STZ groups compared with the control group at 6 months in hippocampus. The total GSK3β showed no significant changes in STZ groups in relation to the controls groups (Figure 5(b)). The ratio between p-GSK3β/GSK3β significantly t(4) = 6.073, p = 0.0009 dropped in the STZ group (Figure 5(c)). In the PFC (Figure 6(a)), p-GSK3 levels did not changes at 6 months in rats with STZ in relation to control group. Total GSK3β and the ratio between p-GSK- 3β/GSK3β were unchanged compared with control (Figure 6(b) and Figure 6(c)). 3.4. STZ Effect on PP2A Levels PP2A levels significantly t(4) = 5.730, p = 0.0012 decreased at 6 months in the Figure 2. Short and long term memory deficit in icv STZ-treated rats. (a) Short (STM; 1.5 h) and long-term memory (LTM; 24 and 48 h) were evaluated one month after STZ icv injection; (b) Memory was evaluated each month during a period of 6 months. Data are plotted as mean ± SEM of conditioned responses. n = 6 - 8. Student’s t test, *p < 0.05 control vs. treated groups. STZ: streptozotocin. T. Ponce-Lopez et al. 20 Figure 3. STZ effect on p-tau in hippocampus. (a) Representative immunoblots of p-tau and total tau, (b) tau phosphorylation and total tau and (c) p-tau/total tau ratio. Six months after injection of STZ-treatment, hippocampal protein extracts were immunoblotted for p-tau (Ser396) (PHF13) and total tau (Tau46). Quantification of tau was normalized against GAPDH. (b) Data expressed as mean ± SEM, n = 4 - 5 animals per group. Significant differences (**p < 0.01) in STZ vs. control group; one-way ANOVA followed by Tukey test. (c) Data are presented as mean ± SEM, n = 4 - 5 animals per group. Student’s t test, *p < 0.05 relative to control group. T. Ponce-Lopez et al. 21 Figure 4. STZ effect on p-tau in prefrontal cortex. (a) Representative immunoblots from a typical experiment, (b) tau phosphorylation and total tau and (c) p-tau/total tau. Six months after injection of STZ prefrontal cortex protein extracts were immunoblotted for p-tau (Ser396) (PHF13) and total tau (Tau46). Quantification of Ser396 p-tau and t-tau were normalized to GAPDH. Data expressed as mean ± SEM, n= 4 - 5 animals per group. Significant differences (*p < 0.05, **p < 0.01) in STZ vs. control group; one-way ANOVA followed by Turkey test. (c) Data are presented as mean ± SEM, n = 4 - 5 animals per group. Student’s t test. T. Ponce-Lopez et al. 22 Figure 5. STZ effect on p-GSK3β in hippocampus. (a) Representative immunoblots of p-GSK3β, (b) Total GSK3β and (c) p-GSK3β/total GSK3β. Six months following STZtreatment, hippocampal protein extracts were immunoblotted for p-GSK3β (Ser9) and total GSK3β (27 C10). Quantification of GSK3β was normalized against GAPDH. (b) Data expressed as mean ± SEM, n = 4 - 5 animals per group. Significant differences (*p < 0.05) in STZ vs. control group; one-way ANOVA followed by Turkey test. (c) Data are presented as mean ± SEM, n = 4 - 5 animals per group. Student’s t test, *p < 0.05 relative to control group. hippocampus of rats with STZ icv compared with the control group (Figure 7(a), Figure 7(b)). PP2A levels did not change at 6 months (Figure 7(c), Figure 7(d)) in the PFC of rats with STZ in relation to control group. 4. Discussion The present study found STM and LTM, and progressive memory impairment following one and six months of icv STZ injection, respectively. It is known that insulin and IR are selectively distributed in the brain, including olfactory bulb, T. Ponce-Lopez et al. 23 hypothalamus, cerebral cortex, amygdala and hippocampus [42] [43]. The expression of IR in cerebral cortex and hippocampus suggests that insulin is involved in memory process [43]. In this context, it should be noted that the levels of IR and mRNA were increased in hippocampus of rat after a spatial memory task, suggesting that insulin might regulate normal memory function [44]. In accordance with this notion, it has been reported in icv STZ rats’ decrease in insulin gene, IR protein and hyperphosphorylated tau protein in the hippocampus and cortex [31], and decrease in IR expression and key proteins of the insulin signaling cascade (e.g. phosphorylation of IRS-1 and Akt) in the CA3 hippocampal region associated with memory damage [45]. Furthermore, the treatment with an Figure 6. STZ effect on p-GSK3β in prefrontal cortex. (a) Representative immunoblots of p-GSK3β, (b) Total GSK3β and (c) p-GSK3β/total GSK3β ratio. Six months following STZ-treatment, prefrontal cortex protein extracts were immunoblotted for p-GSK3β (Ser9) and total GSK3β (27 C10). Quantification of GSK3β was normalized against GAPDH. (b) Data expressed as mean ± SEM, n = 4 - 5 animals per group. Significant differences in STZ vs. control group; one-way ANOVA followed by Tukey test. (c) Data are presented as mean ± SEM, n = 4 - 5 animals per group. Student’s t test, *p < 0.05 relative to control group. T. Ponce-Lopez et al. 24 Figure 7. STZ effect on PP2A levels in hippocampus and prefrontal cortex. (a), (c) Representative immunoblots of PP2A and (b), (d) PP2A levels in hippocampus and prefrontal cortex. Six months following STZ-treatment, protein extracts were immunoblotted for PP2A subunit A (6G3). Quantification of PP2A was normalized against GAPDH. Data are presented as mean ± SEM, n = 4 - 5 animals per group. Student’s t test, *p < 0.01 relative to control group. insulin sensitizer (e.g., pioglitazone) reversed memory deficit in icv STZ rats [30] [38]. On the other hand, insulin dysfunction (e.g., chronic hyperinsulinemia or diabetes mellitus) has a negative impact on memory and cognitive function [46] [47], and clinical studies demonstrate a beneficial effect of intranasal insulin on memory and cognitive function in AD patients [48]. Interestingly, we observed increased tau phosphorylated, decreased p-GSK3β and PP2A in hippocampus but not in PFC. Hyperphosphorylation of tau is considered as one of the typical pathological changes in AD [49]. In the normal brain, balance between phosphorylation and dephosphorylation of tau results in structural and conformational changes that regulate the stability of the cytoskeleton and axonal morphology [50] [51] [52] [53]. During the development of AD and other neurodegenerative disorders, tau is phosphorylated at multiple sites as a result of the imbalance of numerous Ser/Thr kinases (GSK3β) and phosphatases (PP2A) [50] [51] [52] [53], and integrates paired helical filaments (PHF) leading to NFTs and losing their physiological functions [54] [55] [56]. In this study, the increase observed in p-tau was obtained using a monoclonal phosphotau (Ser396). Notably, here the total tau did not change in hippocampus; however, increased p-tau and total tau were observed in PFC. The proportion of p-tau/tau total augmented in hippocampus. Hence, the present results might be due to post-translational modifications (increase in phosphorylation) and not by increase in tau protein in hippocampus, but not in PFC. Regarding GSK3β role, several studies have been shown that insulin could regulate tau phosphorylation in neurons [57] [58] [59]. For instance, binding of insulin to IR induces activating signal transduction cascade of the PI3K pathway T. Ponce-Lopez et al. 25 [60]. The activation of the PI3K, in turn activates protein kinase B (Akt/PKB), then Akt/PKB phosphorylates the GSK3, that results in inactivation of GSK3 [61]. Hence, disruption of IR-PI3K-Akt/PKB signaling cascade leads to the dephosphorylation in Ser9 of GSK3β increases in the activity of GSK3β [31]. Notably, the activated GSK3β isoform has been involved in tau-protein phosphorylation [62]. Moreover, it is recognized that GSK3β isoform is the major kinase that phosphorylates in vivo tau [63] [64], and, that phosphorylates tau in several sites; hyperphosphorylated in PHF, is present in tangles of brain of AD patients [57] [65] [66]. Our results showed decrease in p-GSK3β and p-GSK3β/total GSK3β ratio; this indirectly suggested an increase in the active GSK3β form, therefore, this might explain the consequent increase of p-tau. Consistent with these results, in our laboratory, we found that insufficient inhibitory GSK3β control in STZ-icv rats was demonstrated by a decrease in p-GSK3β levels in the hippocampus, which was associated to STM and LTM impairment (38). Additionally, lithium (GSK3 inhibitor) and pioglitazone (insulin sensitizer) treatment reversed this memory deficit and restored the inhibitory activity of GSK3β in hippocampus [38]. Similarly, other studies have demonstrated that specific inhibition GSK3 activity by lithium prevents hyperphosphorylation of tau, and spatial memory loss resulting from inhibiting the PI3K and PKC [67]. In addition, transgenic mice (Tet/GSK3β) that conditionally over-expresses GSK3 in neurons of the hippocampus and cortex showed spatial memory deficit [68]. Finally, PP2A diminished in the hippocampus but not in PFC; the former was colocalized with tau and microtubuled in the brain [69], and it was apparently the most active enzyme in dephosphorylating the abnormal tau to a normal-like state [9] [63] [70]. In AD brain, both the activity and the mRNA of PP2A are decreased [71] [72] [73]. Thus, the reduction in the activity of the PP2A promotes the hyperphosphorylation of tau and seems to be an important factor in the progression of the AD [71] [72] [73] [74]. Therefore, the abnormal phosphorylation of tau observed herein in hippocampus, might be as a result of decrease in p-GSK3, but also by diminished dephosphorylating activity of PP2A. In agreement with this, the inhibition of PP2A with okadaic acid (OKA), i.e., a potent and selective inhibitor of PP2A and PP1, produces memory impairment in the Morris water maze and oxidative stress, treatment with memantine (10 mg/kg, po) or donepezil (5 mg/kg, po) for 13 days post-OKA, improves memory and reduces oxidative stress [75]. In brief, a failure in IR signaling pathways (PI3K-Akt-GSK3β) and/or insufficient regulation of phosphorylation by PP2A, might be contributing in the regulation of memory functions and tau protein phosphorylation, hence, the insulin signaling played an important role in AD. These data also suggest that inhibition of GSK3β and activation of PP2A could contribute to inhibiting neurofibrillary degeneration and, which represents an important therapeutic target for AD. The hippocampus is perhaps the brain structure that participates the most notably in the regulation of tau phosphorylation than the frontal cortex. The present evidence supports the experimental approach to sporadic AD, based on the insu- T. Ponce-Lopez et al. 26 lin-resistant state induced in the brains of animals following the icv STZ, also, supports the notion that ICV-STZ rat is a suitable model to represent essential features of the SAD and for investigating effective treatments to prevent or reverse memory deficits and neurofibrillary degeneration. References [1] Querfurth, H.W. and La Ferla, F.M. (2010) Alzheimer’s Disease. The New England Journal of Medicine, 362, 329-344. https://doi.org/10.1056/NEJMra0909142 [2] Selkoe, D.J. (2001) Alzheimer’s Disease Results from the Cerebral Accumulation and Cytotoxicity of Amyloid Beta-Protein. Journal of Alzheimer’s Disease, 3, 75-80. [3] Goedert, M. and Spillantini, M.G. (2006) A Century of Alzheimer’s Disease. Science, 314, 777-781. https://doi.org/10.1126/science.1132814 [4] Blennow, K., de Leon, M.J. and Zetterberg, H. (2000) Alzheimer’s Disease. The Lancet, 368, 387-403. https://doi.org/10.1016/S0140-6736(06)69113-7 [5] Buée, L., Bassiére, T. and Buée-Scherrer, V. (2000) Tau Protein Isoforms, Phosphorylation and Role in Neurodegenerative Disorders. Brain Research Review, 33, 95- 130. https://doi.org/10.1016/S0165-0173(00)00019-9 [6] Van Dam, D. and De Deyn, P.P. (2011) Animal Models in the Drug Discovery Pipeline for Alzheimer’s Disease. British Journal of Pharmacology,164, 1285-1300. https://doi.org/10.1111/j.1476-5381.2011.01299.x [7] Planel, E., Tatebayashi, Y., Miyasaka, T., Liu, L., Wang, L., Herman, M., Yu, W.H., Luchsinger, J.A., Wadzinski, B., Duff, K.E. and Takashima, A. (2007) Insulin Dysfunction Induces in Vivo Tau Hyperphosphorylation through Distinct Mechanisms. Journal of Neuroscience, 27, 13635-13648. https://doi.org/10.1523/JNEUROSCI.3949-07.2007 [8] Planel, E., Yasutake, K., Fujita, S.C. and Ishiguro, K. (2001) Inhibition of Protein Phosphatase 2A Overrides Tau Protein Kinase I/Glycogen Synthase Kinase 3beta and Cyclin-Dependant Kinase 5 Inhibition and Results in Tau Hyperphosphorylation in the Hippocampus of Starved Mouse. The Journal of Biological Chemistry, 276, 34298-34306. https://doi.org/10.1074/jbc.M102780200 [9] Wang, J.Z., Grundke-Iqbal, I. and Iqbal, K. (2007) Kinases and Phosphatases and Tau Sites Involved in Alzheimer Neurofibrillary Degeneration. European Journal of Neuroscience, 25, 59-68. https://doi.org/10.1111/j.1460-9568.2006.05226.x [10] Waring, S.C. and Rosenberg, R.N. (2008) Genome-Wide Association Studies in Alzheimer Disease. Archives of Neurology, 65, 329-334. https://doi.org/10.1001/archneur.65.3.329 [11] El Khoury, N.B., Gratuze, M., Papon, M.-A., Bretteville, A. and Planel, E. (2014) Insulin Dysfunction and Tau Pathology. Frontiers in Cellular Neuroscience, 8, 22. https://doi.org/10.3389/fncel.2014.00022 [12] Frolich, L., Blum-Degen, D., Riederer, P. and Hoyer, S. (1999) A Disturbance in the Neuronal Insulin Receptor Signal Transduction in Sporadic Alzheimer’s Disease. Annals of the New York Academy of Sciences, 893, 290-293. https://doi.org/10.1111/j.1749-6632.1999.tb07839.x [13] Gasparini, L., Netzer, W.J., Greengard, P. and Xu, H. (2002) Does Insulin Dysfunction Play a Role in Alzheimer’s Disease? Trends in Pharmacological Sciences, 23, 288-293. https://doi.org/10.1016/S0165-6147(02)02037-0 [14] Craft, S. and Watson, G.S. (2004) Insulin and Neurodegenerative Disease, Shared and Specific Mechanisms. The Lancet Neurology, 3, 169-178. T. Ponce-Lopez et al. 27 https://doi.org/10.1016/S1474-4422(04)00681-7 [15] De La Monte, S.M., Longato, L., Tong, M. and Wands, J.R. (2009) Insulin Resistance and Neurodegeneration, Roles of Obesity, Type 2 Diabetes Mellitus and NonAlcoholic Steatohepatitis. Current Opinion in Investigational Drugs, 10, 1049-1060. [16] De La, M. (2012) Contributions of Brain Insulin Resistance and Deficiency in Amyloid-Related Neurodegeneration in Alzheimer’s Disease. Drugs, 72, 49-66. https://doi.org/10.2165/11597760-000000000-00000 [17] Plaschke, K., Kopitz, J., Siegelin, M., Schliebs, R., Salkovic-Petrisic, M., Riederer, P. and Hoyer, S. (2010) Insulin-Resistant Brain State after Intracerebroventricular Streptozotocin Injection Exacerbates Alzheimer-Like Changes in Tg2576 AbetaPPOverexpressing Mice. Journal of Alzheimer’s Disease, 19, 691-704. [18] Talbot, K., Wang, H.Y., Kazi, H., Han, L.Y., Bakshi, K.P., Stucky, A., Fuino, R.L., Kawaguchi, K.R., Samoyedny, A.J., Wilson, R.S., Arvanitakis, Z., Schneider, J.A., Wolf, B.A., Bennett, D.A., Trojanowski, J.Q. and Arnold, S.E. (2012) Demonstrated Brain Insulin Resistance in Alzheimer’s Disease Patients Is Associated with IGF-1 Resistance, IRS-1 Dysregulation, and Cognitive Decline. Journal of Clinical Investigation, 122, 1316-1338. https://doi.org/10.1172/JCI59903 [19] Neumann, K.F., Rojo, L., Navarrete, L.P., Farías, G. and Maccioni, R.P. (2008) Insulin Resistance and Alzheimer’s Disease, Molecular Links & Clinical Implications. Current Alzheimer Research, 5, 438-447. https://doi.org/10.2174/156720508785908919 [20] Leibson, C.L., Rocca, W.A., Hanson, V.A., Cha, R., Kokmen, E., O’Brien, P.C. and Palumbo, P.J. (1997) Risk of Dementia among Persons with Diabetes Mellitus, a Population-Based Cohort Study. American Journal of Epidemiology, 145, 301-308. https://doi.org/10.1093/oxfordjournals.aje.a009106 [21] Stolk, R.P., Breteler, M.M., Ott, A., Pols, H.A., Lamberts, S.W., Grobbee, D.E. and Hofman, A. (1997) Insulin and Cognitive Function in an Elderly Population. The Rotterdam Study. Diabetes Care, 20, 792-795. https://doi.org/10.2337/diacare.20.5.792 [22] Li, X., Song, D.S. and Leng, S.X. (2015) Link between Type 2 Diabetes and Alzheimer’s Disease, from Epidemiology to Mechanism and Treatment. Clinical Interventions in Aging, 10, 549-560. https://doi.org/10.2147/CIA.S74042 [23] Wallum, B.J., Taborsky, G.J., Porte, D., Figlewicz, D.P., Jacobson, L., Beard, J.C., Ward, W.K. and Dorsa, D. (1987) Cerebrospinal Fluid Insulin Levels Increase during Intravenous Insulin Infusions in Man. The Journal of Clinical Endocrinology & Metabolism, 64, 190-194. https://doi.org/10.1210/jcem-64-1-190 [24] Nitsch, R. and Hoyer, S. (1991) Local Action of the Diabetogenic Drug, Streptozotocin, on Glucose and Energy Metabolism in Rat Brain Cortex. Neuroscience Letters, 128, 199-202. https://doi.org/10.1016/0304-3940(91)90260-Z [25] Hellweg, R., Nitsch, R., Hock, C., Jaksch, M. and Hoyer, S. (1992) Nerve Growth Factor and Choline Acetyltransferase Activity Levels in the Rat Brain Following Experimental Impairment of Cerebral Glucose and Energy Metabolism. Journal of Neuroscience Research, 31, 479-486. https://doi.org/10.1002/jnr.490310310 [26] Plaschke, K. and Hoyer, S. (1993) Action of the Diabetogenic Drug Streptozotocin on Glycolytic and Glycogenolytic Metabolism in Adult Rat Brain Cortex and Hippocampus. International Journal of Developmental Neuroscience, 11, 477-483. https://doi.org/10.1016/0736-5748(93)90021-5 [27] Duelli, R., Schrock, H., Kuschinsky, W. and Hoyer, S. (1994) Intracerebroventricular Injection of Streptozotocin Induces Discrete Local Changes in Cerebral Glucose Utilization in Rats. International Journal of Developmental Neuroscience, 12, 737- T. Ponce-Lopez et al. 28 743. https://doi.org/10.1016/0736-5748(94)90053-1 [28] Sharma, M. and Gupta, Y.K. (2001) Intracerebroventricular Injection of Streptozotocin in Rats Produces Both Oxidative Stress in the Brain and Cognitive Impairment. Life Sciences, 68, 1021-1029. https://doi.org/10.1016/S0024-3205(00)01005-5 [29] Paxinos, G. and Watson, C. (2005) The Rat Brain Stereotaxic Coordinates. Academic Press, Sidney. [30] Pathan, A.R., Viswanad, B., Sonkusare, S.K. and Ramarao, P. (2006) Chronic Administration of Pioglitazone Attenuates Intracerebroventricular Streptozotocin Induced-Memory Impairment in Rats. Life Sciences, 79, 2209-22016. https://doi.org/10.1016/j.lfs.2006.07.018 [31] Grünblatt, E., Salkovic-Petrisic, M., Osmanovic, J., Riederer, P. and Hoyer, S. (2007) Brain Insulin System Dysfunction in Streptozotocin Intracerebroventricularly Treated Rats Generates Hyperphosphorylated Tau Protein. Journal of Neurochemistry, 101, 757-770. https://doi.org/10.1111/j.1471-4159.2006.04368.x [32] Salkovic-Petrisic, M., Tribl, F., Schmidt, M., Hoyer, S. and Riederer, P. (2006) Alzheimer-Like Changes in Protein Kinase B and Glycogen Synthase Kinase-3 in Rat Frontal Cortex and Hippocampus after Damage to the Insulin Signalling Pathway. Journal of Neurochemistry, 96, 1005-1015. https://doi.org/10.1111/j.1471-4159.2005.03637.x [33] Salkovic-Petrisic, M., Osmanovic, J., Grünblatt, E., Riederer, P. and Hoyer, S. (2009) Modeling Sporadic Alzheimer’s Disease, the Insulin Resistant Brain State Generates Multiple Long-Term Morphobiological Abnormalities Including Hyperphosphorylated Tau Protein and Amyloid-Beta. Journal of Alzheimer’s Disease, 18, 729-750. [34] Salkovic-Petrisic, M., Knezovic, A., Siegfred, H. and Riederer, P. (2013) What Have We Learned from the Streptozotocin-Induced Animal Model of Sporadic Alzheimer’s Disease, about the Therapeutic Strategies in Alzheimer’s Research. Journal of Neural Transmission, 120, 233-252. https://doi.org/10.1007/s00702-012-0877-9 [35] Meneses, A. (2003) A Pharmacological Analysis of an Associative Learning Task, 5- HT(1) to 5-HT(7) Receptor Subtypes Function on a Pavlovian/Instrumental a Autoshaped Memory. Learning & Memory, 5, 363-372. https://doi.org/10.1101/lm.60503 [36] Meneses, A. (2002) Tianeptine, 5-HT Uptake Sites and 5-HT (1-7) Receptors Modulate Memory Formation in an Autoshaping Pavlovian/Instrumental Task. Neuroscience & Biobehavioral Reviews, 26, 309-319. https://doi.org/10.1016/S0149-7634(02)00005-2 [37] Meneses, A. (2007) Do Serotonin1-7 Receptors Modulate Short and Long-Term Memory? Neurobiology of Learning and Memory, 87, 561-572. https://doi.org/10.1016/j.nlm.2006.12.005 [38] Ponce-Lopez, T., Liy-Sameron, G., Hong, E. and Meneses, A. (2011) Lithium, Phenserine, Memantine and Pioglitazone Reverse Memory Deficit and Restore PhosphoGSK3β Decreased in Hippocampus in Intracerebroventricular Streptozotocin Induced Memory Deficit Model. Brain Research, 1426, 73-85. https://doi.org/10.1016/j.brainres.2011.09.056 [39] Bradford, M.M. (1976) A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Analytical Biochemistry, 7, 248-254. https://doi.org/10.1016/0003-2697(76)90527-3 [40] Kurien, B.T. and Scofield, R.H. (2003) Protein Blotting, a Review. Journal of Immunological Methods, 274, 1-15. https://doi.org/10.1016/S0022-1759(02)00523-9 [41] Kurien, B.T. and Scofield, R.H. (2006) Western Blotting. Methods, 38, 283-293. https://doi.org/10.1016/j.ymeth.2005.11.007 T. Ponce-Lopez et al. 29 [42] Baskin, D.G., Figlewicz, D.P., Woods, S.C., Porte, D. and Dorsa, D.M. (1987) Insulin in the Brain. Annual Review of Physiology, 49, 335-347. https://doi.org/10.1146/annurev.ph.49.030187.002003 [43] Zhao, W., Chen, H., Xu, H., Moore, E., Meiri, N., Quon, M.J. and Alkon, D.L. (1999) Brain Insulin Receptors and Spatial Memory. Correlated Changes in Gene Expression, Tyrosine Phosphorylation, and Signaling Molecules in the Hippocampus of Water Maze Trained Rats. The Journal of Biological Chemistry, 274, 34893- 34902. https://doi.org/10.1074/jbc.274.49.34893 [44] Zhao, W.Q. and Alkon, D.L. (2001) Role of Insulin and Insulin Receptor in Learning and Memory. Molecular and Cellular Endocrinology, 177, 125-134. https://doi.org/10.1016/S0303-7207(01)00455-5 [45] Agrawal, R., Tyagi, E., Shukla, S. and Nath, C. (2009) A Study of Brain Insulin Receptors, AChE Activity and Oxidative Stress in Rat Model of ICV STZ Induced Dementia. Neuropharmacology, 56, 779-787. https://doi.org/10.1016/j.neuropharm.2009.01.005 [46] Benedict, C., Brooks, S.J., Kullberg, J., Burgos, J., Kempton, M.J., Nordenskjold, R., Nylander, R., Kilander, L., Craft, S., Larsson, E.-M., Johansson, L., Ahlström, H., Lind, L. and Schiöth, H.B. (2012) Impaired Insulin Sensitivity as Indexed by the HOMA Score Is Associated with Deficits in Verbal Fluency and Temporal Lobe Gray Matter Volume in the Elderly. Diabetes Care, 35, 488-494.
Role of GSK3β and PP2A on Regulation of Tau Phosphorylation in Hippocampus and Memory Impairment in ICV-STZ Animal Model of Alzheimer’s Disease Teresa Ponce-Lopez1,2*, Enrique Hong1, Manuel Abascal-Díaz2, Alfredo Meneses1 1 Deparment of Pharmacobiology, CINVESTAV, México City, México 2 Faculty of Health Sciences, Anahuac University, Huixquilucan State of México, Mexico Abstract Intracerebroventricular administration (ICV) of streptozotocin (STZ) in rats has been associated to desensitization of the insulin receptor (IR) and biochemical changes similar to those occurring in Alzheimer’s disease (AD) or older brains, so it has been proposed as a suitable model for studying some of the pathological features of AD sporadic type (SAD). In this study, we investigated the role of glycogen synthase kinase 3β (GSK3β) and protein phosphatase 2A (PP2A) in the regulation of the phosphorylation of tau (p-tau). Results showed that ICV-STZ treated rats had deficits in short- (1.5-h) and long-term (24- and 48-h) memory after one month of ICV-STZ treatment and six months relative to control rats. The memory deficit was associated to increasing [F(3, 12) = 31.48, p < 0.0001] p-tau in the hippocampus but not in prefrontal cortex (PFC). Likewise, STZ reduced phosphorylation of GSK3β (p-GSK3β) and PP2A in hippocampus and PFC, indicating that GSK3β and PP2A contributed to regulation of p-tau. These data supporting the model with ICV-STZ in rat are adequate to study the progressive memory impairment associated to hyperphosphorylation of tau and the cascade of insulin receptor signaling; confirm that phosphatidyl-inositol-3 kinase-protein kinase B (PI3K-PKB/AktGSK3β) and PP2A are involved in the modulation of proteins responsible for the regulation of neurodegeneration in AD. Keywords Memory Deficit, Tau Hyperphosphorylation, GSK3β, PP2A, Streptozotocin, Hippocampus How to cite this paper: Ponce-Lopez, T., Hong, E., Abascal-Díaz, M. and Meneses, A. (2017) Role of GSK3β and PP2A on Regulation of Tau Phosphorylation in Hippocampus and Memory Impairment in ICVSTZ Animal Model of Alzheimer’s Disease. Advances in Alzheimer’s Disease, 6, 13-31. https://doi.org/10.4236/aad.2017.61002 Received: January 21, 2017 Accepted: March 7, 2017 Published: March 10, 2017 Copyright © 2017 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/ Open Access T. Ponce-Lopez et al. 14 1. Introduction Alzheimer’s disease (AD) is the leading cause of dementia and is characterized by progressive memory loss and a gradual decline in cognitive function, eventually leading to premature death of the inpidual, which occurs typically 3 - 9 years after diagnosis [1]. The neuropathological hallmarks of AD are extracellular senile plaques containing aggregates of the β-amyloid peptide (Aβ), and intraneuronal neurofibrillary tangles (NFTs) [2] [3] [4] that consist mainly of intracellular and abnormally phosphorylated tau protein [3] [5]. These pathologic features are accompanied by decreased synaptic density, which eventually leads to widespread neurodegeneration, loss of synapses and failure of neurotransmitter pathways, particularly those of the basal forebrain cholinergic system, especially in the hippocampus and cortex [3] [6]. Tau phosphorylation (ptau) is regulated by numerous Ser/Thr and phosphatases, including glycogen synthase kinase 3β (GSK3β) and protein phosphatase 2A (PP2A); both are considered to be the major kinase and phosphatase in vivo [7] [8] [9]. Several evidences have been associated between insulin dysfunction and AD [10]-[16]. For instance, hyperphosphorylation of tau protein has been associated with brain insulin deficiency or disorder of insulin signal transduction [17]. A post-mortem study showed that the hippocampal formation in AD cases without diabetes, exhibited markedly reduced receptor/insulin receptor substrate1/ phosphatidyli-inositol-3 kinase (IR-IRS1-PI3K) signaling pathway [18]. The other study showed that peripheral hyperinsulinemia correlated with an abnormal removal of Aβ and an increase in tau hyperphosphorylation, consequently of increased GSK3β activity [19]. In fact, hyperinsulinemia and Type II diabetes mellitus are considered risk factors for SAD [16] [20] [21] [22] [23]. Importantly, diabetes animal models suggest that peripheral insulin signaling dysfunction plays a key role in modulating AD pathology. Indeed, streptozotocin (STZ) administration (200 mg/kg i.p.) produced increase of tau phosphorylation in the brain of non-transgenic mice [7]. Likewise, the ICV administration of low STZ doses (1 - 3 mg/kg) reproduces aspects of SAD abnormalities, including decreased glucose utilization in rat cortical regions and hippocampus [24] [25] [26] [27], cholinergic deficits [25], increase in oxidative stress [28] [29] [30], decrease of IR expression and hyperphosphorylated tau protein in the hippocampus [31], and amyloid formation in leptomeningeal vessels [30]. All these changes are associated to memory impairment, and tau pathology, resulting in central insulin dysfunction [30] [31] [32] [33]. Therefore, herein we are testing that ICV-STZ animal model provides key information about the role of brain insulin disruption in AD pathology, hence, studying the effect at six months following icv STZ administration on memory function, phosphorylated levels of tau and GSK3β, and PP2A levels. 2. Methods 2.1. Animals Male Wistar 4-month-old rats weighing 320 - 340 g were used. The animals were T. Ponce-Lopez et al. 15 placed on an inpidual cage after STZ administration during recovery time after surgery (one week). During the experimental phase they were maintained at 22˚C ± 2˚C and 12 h light/dark cycle, with free access to food and water previous to experimental manipulation. The experimental protocol was revised and approved by the Institutional Review Committee (CICUAL; Project No. 047/02) for the use of animal subjects in compliance with the National Institutes of Health Guide for Care and Use of Laboratory Animals (publication No. 85 - 23, revised 1985). 2.2. Surgical Procedure and Icv STZ Administration Adult male Wistar rats were anesthetized with ketamine (75 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.). The animal body was placed in position in the stereotaxic apparatus and in its head a midline sagittal incision was made in the scalp. Then, holes were drilled in the skull on both sides over the lateral ventricles. The following coordinates were used for ICV injection: 0.8 mm posterior to Bregma, 1.5 mm lateral to sagittal suture, 3.6 mm ventral from the surface of the brain [28]. Coordinates for placement of cannulas were determined by using the atlas ofPaxinos & Watson [34]. STZ was dissolved in citrate buffer (CB; pH 4.5; 28, 32) just prior to injection. The STZ group was injected bilaterally with STZ (3 mg/kg) in two pided doses on Days 1 and 3 asreported previously [28]. The concentration of STZ was adjusted so as to deliver 2 μL/ventricule of the solution as described previously [32]. In control group were given icv injection of the same volume of CB on 1 and 3 day as in STZ injected rats. Post-operatively, as above mentioned rats were maintained in an inpidual cage and received free access to food and water. One week previous to the behavioral task body weights were reduced to 85% by gradually reducing food intake until the last experimental day in order to perform the behavioral task. 2.3. Behavioral Protocol 2.3.1. Autoshaping Learning Task In an autoshaping or sign-tracking setting, a hungry animal is placed in a conditioning chamber to find food pellets (unconditioned stimulus [US]) in the foodmagazine and is then given a Pavlovian sequential pairing (stimulus-stimulus [S-S]) of a lighted key or a retractable-illuminated lever (conditioned stimulus [CS]) and food (US). After a number of such presentations, the animal approaches the CS and presents instrumental responses (conditioned response [CR]), such as peck, nose-poke, and lever-press responses. Then, CR or autoshaped responses result from the S-S association and are sustained by response-stimulus (R-S) association [35]. Importantly, within the continued progress of behavioral memory tasks development, a Pavlovian/Instrumental autoshaping (P/I-A) task combines both Pavlovian and instrumental conditioning; which offers the opportunity to study hippocampus-mediated declarative memory and striatum-mediated R-S “habit formation” [36]. Furthermore, except for magazine training, P/IA is almost completely automatized, considerably reducing human intervention. T. Ponce-Lopez et al. 16 It is sensitive to small increases or decreases in various behavioral parameters (i.e., not measuring the same event twice), including sign tracking (i.e., conditioned behavior directed toward the localized retractable and illuminated lever; CS), and goal tracking (i.e., the place where the US is delivered). The latest is quite important, as it allows the study of bidirectional expression of an enhanced or impaired memory formation. P/I-A clearly separates training for testing sessions, and it has been useful to detect changes in memory formation elicited by drugs or aging [35]. 2.3.2. Food Magazine and Autoshaping Training Following one month of the icv STZ administration, inpidually, each rat was placed in an experimental chamber for a habituation period (≈15 min) with access to 50 food-pellets (45 mg each) previously placed into the food magazine. The criterion was that once the animal ate all 50 food-pellets and presented 150 nose-pokes (as measured by a photocell) into the food-magazine, the autoshaping training program was initiated [35] [37]. The autoshaping program had been reported previously [37] [38], and this consisted of discrete trials. A trial began with the presentation of a retractable and illuminated lever for 8 s (conditioned stimulus; CS) followed by a food-pellet (unconditioned stimulus; US) delivery. There was an inter-trial interval time (ITI) of 60 s. When the animal pressed the CS, it was considered a conditioned response (CR), which shortened the trial, retracted the lever, turned off the light, and a US was delivered. The CR increment or decrement was considered an index for enhancement or impairment in memory consolidation, respectively. There was an autoshaping training session (10 trials) lasting nearly 12 min, and three training/testing (20 trails each) sessions, lasting nearly 24 min. All sessions were conducted over three consecutive days. The autoshaping training session is followed by consecutive training/testing sessions at 1.5 h for short-term memory (STM), and 24 and 48 h for long-term memory (LTM). Subsequently, a 20 minute testing session was conducted each month for 6 consecutive months (Figure 1). 2.4. Measured of Brain Proteins 2.4.1. Tissue Preparation As previously reported [38], the rats treated with CB or STZ were sacrificed by decapitation after the autoshaping testing session at 6 months (Figure 1), and their brains were quickly removed, placed on ice in order to dissect the prefrontal cortex and hippocampus (from CA1 area to dentate gyrus) for each group according to Paxinos and Watson, 2005. Hippocampal and prefrontal cortex were homogenized with lysis buffer containing 150 mM NaCl, 50 mMTris-HCl, 5 mM EDTA and protease inhibitors (PMSF, aprotinin, leupeptin and pepstatin). The lysates were centrifuged at 12,000 g per 20 min at 4˚C. The supernatants were removed and kept in new Eppendorf tubes. Samples were frozen and stored at −70˚C until further analysis. Total protein concentration was determined. The amount of protein was assayed according to the method of [39] (Sigma; Cat. No. B6916). For calibration curve bovine serum albumin (BSA) standard was used (Sigma). T. Ponce-Lopez et al. 17 Figure 1. Experimental design. Under anesthesia Wistar rats were bilaterally injected in lateral ventricles with streptozotocin (STZ) or citrate buffer (CB; vehicle) twice on day 1 and 3, after one month short- (STM) and long-term memory (LTM) was assessed in autoshaping, an associative learning task. Subsequently, testing session was conducted each month for 6 consecutive months. The animals were sacrificed at 6 months, the brains were removed and the hippocampus and prefrontal cortex were dissected in order to measure tau protein, GSK3β and PP2A by western blot. ICV: intracerebroventricular. The concentration was determined by measurement of the absorbance at 595 nm. 2.4.2. Western Blot Equal amounts of total protein (20 μg per sample for enzyme for analyses) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis using 10% polyacrylamide gels and transferred to Polyvinylidenedifluoride (PVDF) membrane [40] [41]. The PVDF membranes were blocked by incubation in 5% non-fat milk added to phosphate buffered saline (PBS-T) containing 137 mM de NaCl, 2.7 mM de KCl, 10 mM de Na2HPO4, 2 mM de KH2PO4, pH 7.4, 0.5% Tween 20, 1 h at 22˚C for p-GSK3β and total GSK3β. Blocked blots were incubated on the next day with primary antibody anti-phospho-GSK3β (Ser9) rabbit (1:1000; Cell Signaling, Inc.), total GSK3β (27 C10) rabbit (1:1000; Cell Signaling, Inc.), Tau (Tau46) Mouse (1:10,000; Cell Signaling, Inc.), phospho-tau (Ser396) (PHF13) Mouse; (1:1000, Cell Signaling, Inc.), PP2A subunit A (6G3) rat (1:1000, Cell Signaling, Inc.) and anti-GAPDH clone 6C5 (1:5000; Milipore, Inc.) overnight at 4˚C. After incubation, the membranes were washed three times T. Ponce-Lopez et al. 18 with PBS-T 1% and incubated for 1 h at room temperature with secondary antibody solution anti-rabbit IgG (1:2000) p-GSK3β, total GSK3β, total tau, phospho-tau and PP2A (1:2000; Milipore, Inc.), and anti-mouse IgG (1:10,000, Milipore, Inc.) for GAPDH. The specificity of the signal was checked on control membranes that were not incubated with primary antibody. After washing three times in PBS, the membranes were immunostained using chemiluminescence western blotting detection reagents (Bio Rad) and exposure to an X-ray film. Relative optical density of bands was analyzed using MCID gel analysis software, Imaging Research Inc. 2.4.3. Statistical Analysis The values of the conditioned responses (CR) were expressed as a percentage of the total trials (10 or 20) per session (mean ± SEM) in the autoshaping test, meaning that, e.g., 2 - 3 CR corresponded to 20% - 30%. The CR was expressed as means (±SEM) and was analyzed by Student’s t test (two groups). The n per group was 6 - 8 animals. The p-tau, total tau, p-GSK3β and total GSK3β values were expressed as means (±SEM) and they were analyzed by one way ANOVA (Three or more groups) followed by Tukey test post-hoc. The PP2A was expressed as means (±SEM) and they were analyzed by Student’s t test (two groups). In all comparisons p < 0.05 was considered as significant. The n per group was 4 - 5 animals. The statistical software used was GraphPad Prism version 6.00 for Macintosh, San Diego California USA. 3. Results 3.1. Short and Long Term Memory Deficit in Icv STZ-Treated Rats The results showed that one month after icv injection of STZ did not change conditioned responses (CR) during phase training. Nevertheless, CR significantly t(18) = 3.128, p = 0.0058 decreased during STM (1.5 h) and t(18) = 3.16, p = 0.0054; t(18) = 4.514, p = 0.0003 LTM (24 & 48 h) relative to vehicle animals (Figure 2(a)). Memory was assessed every month for a period of six months, and the memory deficit was significantly maintained t(14) = 8.804, p < 0.0001; t(14) = 2.98, p = 0.0099; t(14) = 5.688, p < 0.0001; t(10) = 5.179, p = 0.0004; t(10) = 5.440, p = 0.0008; t(12) = 4.371, p = 0.0009, respectively (Figure 2(b)). 3.2. STZ Effect on Tau Phosphorylation (P-Tau) Western blot analysis (Figure 3(a)) demonstrated that p-tau levels were significantly [F(3, 12) = 31.48, p < 0.0001] higher in STZ groups than the control group at 6 months in the hippocampus (Figure 3(b)). There were no changes in total tau levels (Figure 3(b)). The ratio between p-tau/tau was significantly t(4) = 2.456, p = 0.0494 increased (Figure 3(c)) in STZ Western blot analysis (Figure 4(a)) in the PFC p-tau levels significantly [F(3, 12) = 21.01, p < 0.0001] augmented in STZ groups compared with controls at 6 months (Figure 4(b)). However, total tau levels significantly [F(3, 8) =9.154, P < 0.0058] also increased in STZ group compared to control (Figure 4(b)). With T. Ponce-Lopez et al. 19 respect to the ratio between p-tau/tau total was not observed significant differences in control and STZ rats (Figure 4(c)). 3.3. STZ Effect on GSK3β Phosphorylation (p-GSK3β) Western blot analysis (Figure 5(a)) showed the values of p-GSK3β levels significantly [F(3, 12) = 31.81, p < 0.0001] decreased in STZ groups compared with the control group at 6 months in hippocampus. The total GSK3β showed no significant changes in STZ groups in relation to the controls groups (Figure 5(b)). The ratio between p-GSK3β/GSK3β significantly t(4) = 6.073, p = 0.0009 dropped in the STZ group (Figure 5(c)). In the PFC (Figure 6(a)), p-GSK3 levels did not changes at 6 months in rats with STZ in relation to control group. Total GSK3β and the ratio between p-GSK- 3β/GSK3β were unchanged compared with control (Figure 6(b) and Figure 6(c)). 3.4. STZ Effect on PP2A Levels PP2A levels significantly t(4) = 5.730, p = 0.0012 decreased at 6 months in the Figure 2. Short and long term memory deficit in icv STZ-treated rats. (a) Short (STM; 1.5 h) and long-term memory (LTM; 24 and 48 h) were evaluated one month after STZ icv injection; (b) Memory was evaluated each month during a period of 6 months. Data are plotted as mean ± SEM of conditioned responses. n = 6 - 8. Student’s t test, *p < 0.05 control vs. treated groups. STZ: streptozotocin. T. Ponce-Lopez et al. 20 Figure 3. STZ effect on p-tau in hippocampus. (a) Representative immunoblots of p-tau and total tau, (b) tau phosphorylation and total tau and (c) p-tau/total tau ratio. Six months after injection of STZ-treatment, hippocampal protein extracts were immunoblotted for p-tau (Ser396) (PHF13) and total tau (Tau46). Quantification of tau was normalized against GAPDH. (b) Data expressed as mean ± SEM, n = 4 - 5 animals per group. Significant differences (**p < 0.01) in STZ vs. control group; one-way ANOVA followed by Tukey test. (c) Data are presented as mean ± SEM, n = 4 - 5 animals per group. Student’s t test, *p < 0.05 relative to control group. T. Ponce-Lopez et al. 21 Figure 4. STZ effect on p-tau in prefrontal cortex. (a) Representative immunoblots from a typical experiment, (b) tau phosphorylation and total tau and (c) p-tau/total tau. Six months after injection of STZ prefrontal cortex protein extracts were immunoblotted for p-tau (Ser396) (PHF13) and total tau (Tau46). Quantification of Ser396 p-tau and t-tau were normalized to GAPDH. Data expressed as mean ± SEM, n= 4 - 5 animals per group. Significant differences (*p < 0.05, **p < 0.01) in STZ vs. control group; one-way ANOVA followed by Turkey test. (c) Data are presented as mean ± SEM, n = 4 - 5 animals per group. Student’s t test. T. Ponce-Lopez et al. 22 Figure 5. STZ effect on p-GSK3β in hippocampus. (a) Representative immunoblots of p-GSK3β, (b) Total GSK3β and (c) p-GSK3β/total GSK3β. Six months following STZtreatment, hippocampal protein extracts were immunoblotted for p-GSK3β (Ser9) and total GSK3β (27 C10). Quantification of GSK3β was normalized against GAPDH. (b) Data expressed as mean ± SEM, n = 4 - 5 animals per group. Significant differences (*p < 0.05) in STZ vs. control group; one-way ANOVA followed by Turkey test. (c) Data are presented as mean ± SEM, n = 4 - 5 animals per group. Student’s t test, *p < 0.05 relative to control group. hippocampus of rats with STZ icv compared with the control group (Figure 7(a), Figure 7(b)). PP2A levels did not change at 6 months (Figure 7(c), Figure 7(d)) in the PFC of rats with STZ in relation to control group. 4. Discussion The present study found STM and LTM, and progressive memory impairment following one and six months of icv STZ injection, respectively. It is known that insulin and IR are selectively distributed in the brain, including olfactory bulb, T. Ponce-Lopez et al. 23 hypothalamus, cerebral cortex, amygdala and hippocampus [42] [43]. The expression of IR in cerebral cortex and hippocampus suggests that insulin is involved in memory process [43]. In this context, it should be noted that the levels of IR and mRNA were increased in hippocampus of rat after a spatial memory task, suggesting that insulin might regulate normal memory function [44]. In accordance with this notion, it has been reported in icv STZ rats’ decrease in insulin gene, IR protein and hyperphosphorylated tau protein in the hippocampus and cortex [31], and decrease in IR expression and key proteins of the insulin signaling cascade (e.g. phosphorylation of IRS-1 and Akt) in the CA3 hippocampal region associated with memory damage [45]. Furthermore, the treatment with an Figure 6. STZ effect on p-GSK3β in prefrontal cortex. (a) Representative immunoblots of p-GSK3β, (b) Total GSK3β and (c) p-GSK3β/total GSK3β ratio. Six months following STZ-treatment, prefrontal cortex protein extracts were immunoblotted for p-GSK3β (Ser9) and total GSK3β (27 C10). Quantification of GSK3β was normalized against GAPDH. (b) Data expressed as mean ± SEM, n = 4 - 5 animals per group. Significant differences in STZ vs. control group; one-way ANOVA followed by Tukey test. (c) Data are presented as mean ± SEM, n = 4 - 5 animals per group. Student’s t test, *p < 0.05 relative to control group. T. Ponce-Lopez et al. 24 Figure 7. STZ effect on PP2A levels in hippocampus and prefrontal cortex. (a), (c) Representative immunoblots of PP2A and (b), (d) PP2A levels in hippocampus and prefrontal cortex. Six months following STZ-treatment, protein extracts were immunoblotted for PP2A subunit A (6G3). Quantification of PP2A was normalized against GAPDH. Data are presented as mean ± SEM, n = 4 - 5 animals per group. Student’s t test, *p < 0.01 relative to control group. insulin sensitizer (e.g., pioglitazone) reversed memory deficit in icv STZ rats [30] [38]. On the other hand, insulin dysfunction (e.g., chronic hyperinsulinemia or diabetes mellitus) has a negative impact on memory and cognitive function [46] [47], and clinical studies demonstrate a beneficial effect of intranasal insulin on memory and cognitive function in AD patients [48]. Interestingly, we observed increased tau phosphorylated, decreased p-GSK3β and PP2A in hippocampus but not in PFC. Hyperphosphorylation of tau is considered as one of the typical pathological changes in AD [49]. In the normal brain, balance between phosphorylation and dephosphorylation of tau results in structural and conformational changes that regulate the stability of the cytoskeleton and axonal morphology [50] [51] [52] [53]. During the development of AD and other neurodegenerative disorders, tau is phosphorylated at multiple sites as a result of the imbalance of numerous Ser/Thr kinases (GSK3β) and phosphatases (PP2A) [50] [51] [52] [53], and integrates paired helical filaments (PHF) leading to NFTs and losing their physiological functions [54] [55] [56]. In this study, the increase observed in p-tau was obtained using a monoclonal phosphotau (Ser396). Notably, here the total tau did not change in hippocampus; however, increased p-tau and total tau were observed in PFC. The proportion of p-tau/tau total augmented in hippocampus. Hence, the present results might be due to post-translational modifications (increase in phosphorylation) and not by increase in tau protein in hippocampus, but not in PFC. Regarding GSK3β role, several studies have been shown that insulin could regulate tau phosphorylation in neurons [57] [58] [59]. For instance, binding of insulin to IR induces activating signal transduction cascade of the PI3K pathway T. Ponce-Lopez et al. 25 [60]. The activation of the PI3K, in turn activates protein kinase B (Akt/PKB), then Akt/PKB phosphorylates the GSK3, that results in inactivation of GSK3 [61]. Hence, disruption of IR-PI3K-Akt/PKB signaling cascade leads to the dephosphorylation in Ser9 of GSK3β increases in the activity of GSK3β [31]. Notably, the activated GSK3β isoform has been involved in tau-protein phosphorylation [62]. Moreover, it is recognized that GSK3β isoform is the major kinase that phosphorylates in vivo tau [63] [64], and, that phosphorylates tau in several sites; hyperphosphorylated in PHF, is present in tangles of brain of AD patients [57] [65] [66]. Our results showed decrease in p-GSK3β and p-GSK3β/total GSK3β ratio; this indirectly suggested an increase in the active GSK3β form, therefore, this might explain the consequent increase of p-tau. Consistent with these results, in our laboratory, we found that insufficient inhibitory GSK3β control in STZ-icv rats was demonstrated by a decrease in p-GSK3β levels in the hippocampus, which was associated to STM and LTM impairment (38). Additionally, lithium (GSK3 inhibitor) and pioglitazone (insulin sensitizer) treatment reversed this memory deficit and restored the inhibitory activity of GSK3β in hippocampus [38]. Similarly, other studies have demonstrated that specific inhibition GSK3 activity by lithium prevents hyperphosphorylation of tau, and spatial memory loss resulting from inhibiting the PI3K and PKC [67]. In addition, transgenic mice (Tet/GSK3β) that conditionally over-expresses GSK3 in neurons of the hippocampus and cortex showed spatial memory deficit [68]. Finally, PP2A diminished in the hippocampus but not in PFC; the former was colocalized with tau and microtubuled in the brain [69], and it was apparently the most active enzyme in dephosphorylating the abnormal tau to a normal-like state [9] [63] [70]. In AD brain, both the activity and the mRNA of PP2A are decreased [71] [72] [73]. Thus, the reduction in the activity of the PP2A promotes the hyperphosphorylation of tau and seems to be an important factor in the progression of the AD [71] [72] [73] [74]. Therefore, the abnormal phosphorylation of tau observed herein in hippocampus, might be as a result of decrease in p-GSK3, but also by diminished dephosphorylating activity of PP2A. In agreement with this, the inhibition of PP2A with okadaic acid (OKA), i.e., a potent and selective inhibitor of PP2A and PP1, produces memory impairment in the Morris water maze and oxidative stress, treatment with memantine (10 mg/kg, po) or donepezil (5 mg/kg, po) for 13 days post-OKA, improves memory and reduces oxidative stress [75]. In brief, a failure in IR signaling pathways (PI3K-Akt-GSK3β) and/or insufficient regulation of phosphorylation by PP2A, might be contributing in the regulation of memory functions and tau protein phosphorylation, hence, the insulin signaling played an important role in AD. These data also suggest that inhibition of GSK3β and activation of PP2A could contribute to inhibiting neurofibrillary degeneration and, which represents an important therapeutic target for AD. The hippocampus is perhaps the brain structure that participates the most notably in the regulation of tau phosphorylation than the frontal cortex. The present evidence supports the experimental approach to sporadic AD, based on the insu- T. Ponce-Lopez et al. 26 lin-resistant state induced in the brains of animals following the icv STZ, also, supports the notion that ICV-STZ rat is a suitable model to represent essential features of the SAD and for investigating effective treatments to prevent or reverse memory deficits and neurofibrillary degeneration. References [1] Querfurth, H.W. and La Ferla, F.M. (2010) Alzheimer’s Disease. The New England Journal of Medicine, 362, 329-344. https://doi.org/10.1056/NEJMra0909142 [2] Selkoe, D.J. (2001) Alzheimer’s Disease Results from the Cerebral Accumulation and Cytotoxicity of Amyloid Beta-Protein. Journal of Alzheimer’s Disease, 3, 75-80. [3] Goedert, M. and Spillantini, M.G. (2006) A Century of Alzheimer’s Disease. Science, 314, 777-781. https://doi.org/10.1126/science.1132814 [4] Blennow, K., de Leon, M.J. and Zetterberg, H. (2000) Alzheimer’s Disease. The Lancet, 368, 387-403. https://doi.org/10.1016/S0140-6736(06)69113-7 [5] Buée, L., Bassiére, T. and Buée-Scherrer, V. (2000) Tau Protein Isoforms, Phosphorylation and Role in Neurodegenerative Disorders. Brain Research Review, 33, 95- 130. https://doi.org/10.1016/S0165-0173(00)00019-9 [6] Van Dam, D. and De Deyn, P.P. (2011) Animal Models in the Drug Discovery Pipeline for Alzheimer’s Disease. British Journal of Pharmacology,164, 1285-1300. https://doi.org/10.1111/j.1476-5381.2011.01299.x [7] Planel, E., Tatebayashi, Y., Miyasaka, T., Liu, L., Wang, L., Herman, M., Yu, W.H., Luchsinger, J.A., Wadzinski, B., Duff, K.E. and Takashima, A. (2007) Insulin Dysfunction Induces in Vivo Tau Hyperphosphorylation through Distinct Mechanisms. Journal of Neuroscience, 27, 13635-13648. https://doi.org/10.1523/JNEUROSCI.3949-07.2007 [8] Planel, E., Yasutake, K., Fujita, S.C. and Ishiguro, K. (2001) Inhibition of Protein Phosphatase 2A Overrides Tau Protein Kinase I/Glycogen Synthase Kinase 3beta and Cyclin-Dependant Kinase 5 Inhibition and Results in Tau Hyperphosphorylation in the Hippocampus of Starved Mouse. The Journal of Biological Chemistry, 276, 34298-34306. https://doi.org/10.1074/jbc.M102780200 [9] Wang, J.Z., Grundke-Iqbal, I. and Iqbal, K. (2007) Kinases and Phosphatases and Tau Sites Involved in Alzheimer Neurofibrillary Degeneration. European Journal of Neuroscience, 25, 59-68. https://doi.org/10.1111/j.1460-9568.2006.05226.x [10] Waring, S.C. and Rosenberg, R.N. (2008) Genome-Wide Association Studies in Alzheimer Disease. Archives of Neurology, 65, 329-334. https://doi.org/10.1001/archneur.65.3.329 [11] El Khoury, N.B., Gratuze, M., Papon, M.-A., Bretteville, A. and Planel, E. (2014) Insulin Dysfunction and Tau Pathology. Frontiers in Cellular Neuroscience, 8, 22. https://doi.org/10.3389/fncel.2014.00022 [12] Frolich, L., Blum-Degen, D., Riederer, P. and Hoyer, S. (1999) A Disturbance in the Neuronal Insulin Receptor Signal Transduction in Sporadic Alzheimer’s Disease. Annals of the New York Academy of Sciences, 893, 290-293. https://doi.org/10.1111/j.1749-6632.1999.tb07839.x [13] Gasparini, L., Netzer, W.J., Greengard, P. and Xu, H. (2002) Does Insulin Dysfunction Play a Role in Alzheimer’s Disease? Trends in Pharmacological Sciences, 23, 288-293. https://doi.org/10.1016/S0165-6147(02)02037-0 [14] Craft, S. and Watson, G.S. (2004) Insulin and Neurodegenerative Disease, Shared and Specific Mechanisms. The Lancet Neurology, 3, 169-178. T. Ponce-Lopez et al. 27 https://doi.org/10.1016/S1474-4422(04)00681-7 [15] De La Monte, S.M., Longato, L., Tong, M. and Wands, J.R. (2009) Insulin Resistance and Neurodegeneration, Roles of Obesity, Type 2 Diabetes Mellitus and NonAlcoholic Steatohepatitis. Current Opinion in Investigational Drugs, 10, 1049-1060. [16] De La, M. (2012) Contributions of Brain Insulin Resistance and Deficiency in Amyloid-Related Neurodegeneration in Alzheimer’s Disease. Drugs, 72, 49-66. https://doi.org/10.2165/11597760-000000000-00000 [17] Plaschke, K., Kopitz, J., Siegelin, M., Schliebs, R., Salkovic-Petrisic, M., Riederer, P. and Hoyer, S. (2010) Insulin-Resistant Brain State after Intracerebroventricular Streptozotocin Injection Exacerbates Alzheimer-Like Changes in Tg2576 AbetaPPOverexpressing Mice. Journal of Alzheimer’s Disease, 19, 691-704. [18] Talbot, K., Wang, H.Y., Kazi, H., Han, L.Y., Bakshi, K.P., Stucky, A., Fuino, R.L., Kawaguchi, K.R., Samoyedny, A.J., Wilson, R.S., Arvanitakis, Z., Schneider, J.A., Wolf, B.A., Bennett, D.A., Trojanowski, J.Q. and Arnold, S.E. (2012) Demonstrated Brain Insulin Resistance in Alzheimer’s Disease Patients Is Associated with IGF-1 Resistance, IRS-1 Dysregulation, and Cognitive Decline. Journal of Clinical Investigation, 122, 1316-1338. https://doi.org/10.1172/JCI59903 [19] Neumann, K.F., Rojo, L., Navarrete, L.P., Farías, G. and Maccioni, R.P. (2008) Insulin Resistance and Alzheimer’s Disease, Molecular Links & Clinical Implications. Current Alzheimer Research, 5, 438-447. https://doi.org/10.2174/156720508785908919 [20] Leibson, C.L., Rocca, W.A., Hanson, V.A., Cha, R., Kokmen, E., O’Brien, P.C. and Palumbo, P.J. (1997) Risk of Dementia among Persons with Diabetes Mellitus, a Population-Based Cohort Study. American Journal of Epidemiology, 145, 301-308. https://doi.org/10.1093/oxfordjournals.aje.a009106 [21] Stolk, R.P., Breteler, M.M., Ott, A., Pols, H.A., Lamberts, S.W., Grobbee, D.E. and Hofman, A. (1997) Insulin and Cognitive Function in an Elderly Population. The Rotterdam Study. Diabetes Care, 20, 792-795. https://doi.org/10.2337/diacare.20.5.792 [22] Li, X., Song, D.S. and Leng, S.X. (2015) Link between Type 2 Diabetes and Alzheimer’s Disease, from Epidemiology to Mechanism and Treatment. Clinical Interventions in Aging, 10, 549-560. https://doi.org/10.2147/CIA.S74042 [23] Wallum, B.J., Taborsky, G.J., Porte, D., Figlewicz, D.P., Jacobson, L., Beard, J.C., Ward, W.K. and Dorsa, D. (1987) Cerebrospinal Fluid Insulin Levels Increase during Intravenous Insulin Infusions in Man. The Journal of Clinical Endocrinology & Metabolism, 64, 190-194. https://doi.org/10.1210/jcem-64-1-190 [24] Nitsch, R. and Hoyer, S. (1991) Local Action of the Diabetogenic Drug, Streptozotocin, on Glucose and Energy Metabolism in Rat Brain Cortex. Neuroscience Letters, 128, 199-202. https://doi.org/10.1016/0304-3940(91)90260-Z [25] Hellweg, R., Nitsch, R., Hock, C., Jaksch, M. and Hoyer, S. (1992) Nerve Growth Factor and Choline Acetyltransferase Activity Levels in the Rat Brain Following Experimental Impairment of Cerebral Glucose and Energy Metabolism. Journal of Neuroscience Research, 31, 479-486. https://doi.org/10.1002/jnr.490310310 [26] Plaschke, K. and Hoyer, S. (1993) Action of the Diabetogenic Drug Streptozotocin on Glycolytic and Glycogenolytic Metabolism in Adult Rat Brain Cortex and Hippocampus. International Journal of Developmental Neuroscience, 11, 477-483. https://doi.org/10.1016/0736-5748(93)90021-5 [27] Duelli, R., Schrock, H., Kuschinsky, W. and Hoyer, S. (1994) Intracerebroventricular Injection of Streptozotocin Induces Discrete Local Changes in Cerebral Glucose Utilization in Rats. International Journal of Developmental Neuroscience, 12, 737- T. Ponce-Lopez et al. 28 743. https://doi.org/10.1016/0736-5748(94)90053-1 [28] Sharma, M. and Gupta, Y.K. (2001) Intracerebroventricular Injection of Streptozotocin in Rats Produces Both Oxidative Stress in the Brain and Cognitive Impairment. Life Sciences, 68, 1021-1029. https://doi.org/10.1016/S0024-3205(00)01005-5 [29] Paxinos, G. and Watson, C. (2005) The Rat Brain Stereotaxic Coordinates. Academic Press, Sidney. [30] Pathan, A.R., Viswanad, B., Sonkusare, S.K. and Ramarao, P. (2006) Chronic Administration of Pioglitazone Attenuates Intracerebroventricular Streptozotocin Induced-Memory Impairment in Rats. Life Sciences, 79, 2209-22016. https://doi.org/10.1016/j.lfs.2006.07.018 [31] Grünblatt, E., Salkovic-Petrisic, M., Osmanovic, J., Riederer, P. and Hoyer, S. (2007) Brain Insulin System Dysfunction in Streptozotocin Intracerebroventricularly Treated Rats Generates Hyperphosphorylated Tau Protein. Journal of Neurochemistry, 101, 757-770. https://doi.org/10.1111/j.1471-4159.2006.04368.x [32] Salkovic-Petrisic, M., Tribl, F., Schmidt, M., Hoyer, S. and Riederer, P. (2006) Alzheimer-Like Changes in Protein Kinase B and Glycogen Synthase Kinase-3 in Rat Frontal Cortex and Hippocampus after Damage to the Insulin Signalling Pathway. Journal of Neurochemistry, 96, 1005-1015. https://doi.org/10.1111/j.1471-4159.2005.03637.x [33] Salkovic-Petrisic, M., Osmanovic, J., Grünblatt, E., Riederer, P. and Hoyer, S. (2009) Modeling Sporadic Alzheimer’s Disease, the Insulin Resistant Brain State Generates Multiple Long-Term Morphobiological Abnormalities Including Hyperphosphorylated Tau Protein and Amyloid-Beta. Journal of Alzheimer’s Disease, 18, 729-750. [34] Salkovic-Petrisic, M., Knezovic, A., Siegfred, H. and Riederer, P. (2013) What Have We Learned from the Streptozotocin-Induced Animal Model of Sporadic Alzheimer’s Disease, about the Therapeutic Strategies in Alzheimer’s Research. Journal of Neural Transmission, 120, 233-252. https://doi.org/10.1007/s00702-012-0877-9 [35] Meneses, A. (2003) A Pharmacological Analysis of an Associative Learning Task, 5- HT(1) to 5-HT(7) Receptor Subtypes Function on a Pavlovian/Instrumental a Autoshaped Memory. Learning & Memory, 5, 363-372. https://doi.org/10.1101/lm.60503 [36] Meneses, A. (2002) Tianeptine, 5-HT Uptake Sites and 5-HT (1-7) Receptors Modulate Memory Formation in an Autoshaping Pavlovian/Instrumental Task. Neuroscience & Biobehavioral Reviews, 26, 309-319. https://doi.org/10.1016/S0149-7634(02)00005-2 [37] Meneses, A. (2007) Do Serotonin1-7 Receptors Modulate Short and Long-Term Memory? Neurobiology of Learning and Memory, 87, 561-572. https://doi.org/10.1016/j.nlm.2006.12.005 [38] Ponce-Lopez, T., Liy-Sameron, G., Hong, E. and Meneses, A. (2011) Lithium, Phenserine, Memantine and Pioglitazone Reverse Memory Deficit and Restore PhosphoGSK3β Decreased in Hippocampus in Intracerebroventricular Streptozotocin Induced Memory Deficit Model. Brain Research, 1426, 73-85. https://doi.org/10.1016/j.brainres.2011.09.056 [39] Bradford, M.M. (1976) A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Analytical Biochemistry, 7, 248-254. https://doi.org/10.1016/0003-2697(76)90527-3 [40] Kurien, B.T. and Scofield, R.H. (2003) Protein Blotting, a Review. Journal of Immunological Methods, 274, 1-15. https://doi.org/10.1016/S0022-1759(02)00523-9 [41] Kurien, B.T. and Scofield, R.H. (2006) Western Blotting. Methods, 38, 283-293. https://doi.org/10.1016/j.ymeth.2005.11.007 T. Ponce-Lopez et al. 29 [42] Baskin, D.G., Figlewicz, D.P., Woods, S.C., Porte, D. and Dorsa, D.M. (1987) Insulin in the Brain. Annual Review of Physiology, 49, 335-347. https://doi.org/10.1146/annurev.ph.49.030187.002003 [43] Zhao, W., Chen, H., Xu, H., Moore, E., Meiri, N., Quon, M.J. and Alkon, D.L. (1999) Brain Insulin Receptors and Spatial Memory. Correlated Changes in Gene Expression, Tyrosine Phosphorylation, and Signaling Molecules in the Hippocampus of Water Maze Trained Rats. The Journal of Biological Chemistry, 274, 34893- 34902. https://doi.org/10.1074/jbc.274.49.34893 [44] Zhao, W.Q. and Alkon, D.L. (2001) Role of Insulin and Insulin Receptor in Learning and Memory. Molecular and Cellular Endocrinology, 177, 125-134. https://doi.org/10.1016/S0303-7207(01)00455-5 [45] Agrawal, R., Tyagi, E., Shukla, S. and Nath, C. (2009) A Study of Brain Insulin Receptors, AChE Activity and Oxidative Stress in Rat Model of ICV STZ Induced Dementia. Neuropharmacology, 56, 779-787. https://doi.org/10.1016/j.neuropharm.2009.01.005 [46] Benedict, C., Brooks, S.J., Kullberg, J., Burgos, J., Kempton, M.J., Nordenskjold, R., Nylander, R., Kilander, L., Craft, S., Larsson, E.-M., Johansson, L., Ahlström, H., Lind, L. and Schiöth, H.B. (2012) Impaired Insulin Sensitivity as Indexed by the HOMA Score Is Associated with Deficits in Verbal Fluency and Temporal Lobe Gray Matter Volume in the Elderly. Diabetes Care, 35, 488-494.
